Among compounds containing a closed conjugated system of π-electrons, aromatic compounds are interesting. Despite the high degree of unsaturation, aromatic compounds are resistant to oxidizing agents and temperature, and they are more prone to undergo substitution reactions rather than addition reactions. These compounds have increased thermodynamic stability compared to conjugated open-chain systems. The tendency of some cyclic compounds to become aromatic under favorable conditions is also known.
Aromatic compounds primarily include benzene and substances similar to it. But they can also have a significantly different structure. A closed chain can consist not only of 12 C (carbocycles), but also contain heteroatoms (heterocycles). A single closed system of π-electrons can be formed through both π,π- and p,π-conjugation. The set of characteristic properties of conjugate systems was united by the concept aromaticity. In 1865, F.A. Kekule proposed to describe benzene using two structures, between which the benzene molecule oscillates. But Kekule's individual structure cannot explain the symmetry and characteristic reactivity of benzene. Benzene is a flat regular hexagon with bond angles of 120°. All 12 C-12 C bonds are equivalent, their length is 0.139 nm, i.e., it is intermediate between the lengths of single and double bonds. All 12 C are in sp 2 hybridization, and all σ C-C and C-H bonds are in the same plane .
Each 12 C in a benzene molecule has one unhybridized p-orbital. Six of these orbitals are located perpendicular to the flat σ-skeleton and parallel to each other. When they overlap each other, a single π-electron cloud is formed, i.e., circular conjugation occurs. The π-Electron density is uniformly distributed throughout the cyclic system.

Determining the heats of combustion or hydrogenation of cyclic compounds and comparing experimental values with calculated values, based on the assumption that the compound contains only isolated double bonds, is one piece of evidence for aromaticity. When cyclohexene is hydrogenated to cyclohexane, 120 kJ/mol of heat is released.
If we imagine benzene with a Kekulé structure with three double bonds, then the heat of hydrogenation of benzene should be three times greater than the heat of hydrogenation of cyclohexene:

The experimentally determined value is much smaller. Therefore, benzene has less energy than the hypothetical cyclohexatriene. 151 kJ/mol - empirical conjugation energy (delocalization energy). For benzene, the conjugation energy is an order of magnitude higher than for 1,3-butadiene. To disrupt the aromatic system of benzene, you need to expend an amount of energy equal to this value.
Aromaticity criteria. Based on theoretical calculations and experimental studies of cyclic conjugated systems, it was found that a compound is aromatic if it has:
flat cyclic σ-skeleton;
conjugated closed π-electron system, covering all atoms of the ring and containing 4p + 2π- electrons, where P = 0, 1, 2, 3, etc. – Hückel's rule.
Aromaticity criteria allow one to distinguish conjugated aromatic systems from all others. Benzene contains a sextet of π electrons and follows Hückel's rule at P = 1.
Condensed aromatic systems. Hückel's rule was formulated for planar monocyclic systems. But it can also be applied to planar condensed systems in which there are no atoms that are common to more than two cycles. Such systems include polynuclear aromatic hydrocarbons - naphthalene, anthracene, phenanthrene:

In these compounds, all carbon atoms are in a state of sp 2 hybridization, the cyclic σ skeleton is flat, the π electron cloud covers all the carbon atoms of the cycles, and the number of π electrons obeys Hückel’s rule. In condensed arenes, the electron density is not completely equalized, and they are less thermodynamically stable.
Many aromatic polycyclic hydrocarbons have carcinogenic properties and are being intensively studied in connection with the problems of cancer occurrence and prevention. Some carcinogenic aromatic compounds are found in tobacco smoke.
Non-benzenoid aromatic compounds. There are cyclic conjugated systems that do not contain six-membered rings, but meet the criteria for aromaticity and have aromatic properties. Hückel's rule does not limit the manifestation of aromaticity to neutral particles only. Carbanions and carbocations can be aromatic.
The neutral cyclopentadiene molecule is not aromatic, (one 12 C in sp 3 hybridization and has no R- AO, the cycle is not flat). The hydrogen atoms of the methylene group are very mobile. When cyclopentadiene is exposed to sodium in tetrahydrofuran or sodium hydride in 1,2-dimethoxyethane, a proton is removed and a cyclopentadienide ion is formed:

After the C-H bond is broken, 12 C is left with two electrons. Now all carbon atoms are in the sp 2 hybrid state, the molecule has a flat cyclic σ skeleton and a single closed conjugated system containing six π electrons in five p orbitals. This meets all criteria for aromaticity. To reflect the uniform distribution of “-” charge, the cyclopentadienide ion is depicted as a structure with a circle and a minus sign in the circle:

The cyclopentadienide ion is a π-excess system that acts as a donor of electron density in relation to atoms or molecules with vacant orbitals. It forms metallocenes (ferrocene) with metal ions. In ferrocene, the iron ion is equidistant between two parallel planes of cyclopentadienide ions—a “sandwich structure.” A ferrocene derivative, ferrocerone, stimulates hematopoietic processes and is used for anemia.
Cycloheptatriene is a cyclic system containing seven 12 C and 6 π electrons. And in this case, the 12 C of the methylene group is in the state of sp 3 hybridization and does not have a p orbital. When hydrogen is removed from the methylene group in the form of a hydride ion, a cycloheptatrienyl cation (tropylium cation) is formed:

In the tropylium cation, the seventh p-orbital is vacant and overlaps with neighboring p-orbitals to form a single conjugated system. It meets the criteria for aromaticity. The positive charge is evenly distributed throughout the system. The seven-membered ring lies in the same plane, the C-C distances are 0.140 nm. The seven-membered tropolone aromatic system is common in nature. Some tropolone derivatives are natural antibiotics - fungicides.

Another example of non-benzenoid aromatic compounds is azulene. It is a hydrocarbon containing fused seven-membered and five-membered rings. Each of the 10 12 C is in a state of sp 2 hybridization. A single conjugated system contains 10 π electrons. Azulene is aromatic and has a high stabilization energy (180 kJ/mol). Unlike other aromatic hydrocarbons, azulene (I) has a dipole moment. The presence of a dipole moment suggests that a significant contribution to the structure of azulene is made by structure (II), in which one ring is a cyclopentadienide ion, and the second is an aromatic tropylium cation:

6801 0
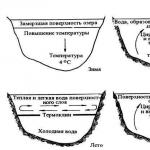
s-elements are elements of groups I and II (+ He, located in group 0 (VIII) of the periodic table). All of them, except H and He, are metals. Group I metals are called alkaline because they react with water to form alkalis. Group II metals, except Be, are called alkaline earth. The term "alkaline earth" refers to the oxides of these metals. These oxides react with water to form alkalis.
S-elements are characterized by the configuration of valence electrons ns 1 (group 1) or ns 2 (group 2). The most important chemical property of group 1 metals is the ability to form positive singly charged cations. High values of the redox potentials of metals of groups I and II indicate their significant reactivity, therefore, under normal conditions they are found only in the form of ions. These ions, depending on the pH, can be found in free form or in the form of complexes, most often with water, carbon dioxide, and halogen ions.
Alkali metals react with water, releasing a large amount of heat ( exothermic reaction). As the atomic number increases, the reactivity increases so much that, starting with K, the H2 formed in the reaction ignites, often explosively. The significant reducing ability of alkali metals is explained by their ease of electron donation.
Table 1. Metals with electron configuration s
|
1(main) |
2 (main) |
|
|
N |
|
|
|
Li |
Be |
|
|
Na |
Mg |
|
|
TO |
Ca |
|
|
Rb |
Sr |
|
|
Cs |
Ba |
|
|
Fr |
Ra |
|
The outer shell of the atoms of all s-metals contains one (for elements of group I - Li, Na, K, Rb, Cs, Fr) or two (for elements of group II - Be, Mg, Ca, Sr, Ba, Ra) electron, very easily separated to form ions, which are similar in configuration to the electron shells of noble gases. Isotopes of all alkali metals contain uncompensated nuclear spin, that is, they are paramagnetic. In whole blood, these isotopes are distributed according to the following rule: the larger the radius of the ion, the higher its content in blood cells(Table 6).
Table 2. The relationship between the distribution coefficient K p and the radius of alkali metal ions
|
Atomic Number | |||
Since group I metal ions are large but do not contain d-electrons, they form complex compounds weakly, and their ability to form complexes decreases with increasing atomic number. If chelate complexes do form, their ligands most often turn out to be oxygen-containing.
Group II metals form complex compounds more easily. As a rule, the ligands in this case are strong complexing agents with oxygen and nitrogen electron-supporting atoms. For example, the chelate ligand EDTA (Fig. 1) is often used for titrimetric determination of ion concentration Sa 2+ and Mg 2+ when analyzing water hardness.
Rice. 1. Complex of ethylenediaminetetraacetic acid (EDTA, "trilon B") with Ca 2+
Concluding the description of s-metals, we note the features of the properties Li And Be, explained by the location of these elements in the upper part of groups I and II, respectively, of the Periodic Table:
1) high compared to other members of the ionization energy groups. This explains the covalent, rather than ionic, nature of the compounds formed by these elements;
2) smaller ion radii than other group members, causing:
a) high charge density, therefore, increased polarizing ability, which is manifested in the increased covalency of their compounds;
b) increased lattice energies of compounds of these elements, explaining their reduced solubility;
3) lower electropositivity compared to other members of the groups. However Li has a high redox potential due to greater hydration energy.
For biochemistry and medicine it is essential that the properties Li and its compounds are in many ways similar to the properties Mg and its connections. It is known that Mg 2+ plays a very important role in metabolism in a living organism, activating ATP and many important enzymes.
Similar diagonal the relationship between elements in the periodic table is also noted for the pair Be And Al with their connections though Be(II group) according to the electronic configuration belongs to the s-elements, and Al(III group) - to p-elements. In addition, the diagonal relationship is described for the pair B - Si and for ions Na + — Sa 2+ — Y 3+ .
The biological role of s-elements is very great. Ions Na + , TO + , Sa 2+ , Mg 2+ , Cl- And NS O 3 - found in all biological fluids. In particular, Na+ and Cl- Contained in large quantities in blood plasma and provide its osmotic pressure. In nerve cells, sudden changes in ion concentration Na+ and TO+ cause electrical impulses that transmit signals in the nervous system.
A sodium pump operates in the plasmalemma of nerve cells, providing a high concentration of ions TO+ inside nerve cells, and ions Na+ - in the intercellular fluid. When ions TO+ diffuse outside the cell, a potential difference arises on its plasmalemma, caused by an excess of ions inside the cell Cl- having a negative charge. After stimulation of the cell, ions begin to pass through the plasmalemma Na+ , as a result of which the sign of the potential difference changes to the opposite. After this, the electrical impulse begins to spread.
One of the manifestations of homeostasis is also considered to be the fact that sodium chloride is part of the sweat fluid, the release of which helps to cool the body.
Medical bioinorganics. G.K. Barashkov
4.D, L - System for naming stereoisomers.
In a number of cases, they prefer to use not the R, S - system for designating the absolute configuration, but another, D, L - system. The choice of D or L isomer design is based on the specific location reger group in the Fischer projection . D ,L-nomenclature is widely used in the names of -amino, -hydroxy acids and carbohydrates.
According to this system, the L-configuration is assigned to a stereosomer, in which in the Fischer projection the reference group is located to the left of the vertical line (from the Latin “laevus” - left). Accordingly, if the reference group located in the Fischer projection on the right, the stereoisomer has a D configuration (from the Latin “dexter” - right):

Of course, we must remember that in the Fischer projection the most oxidized carbon atom is located at the top (that is, the COOH group in amino- and hydroxyl acids and the CH=O group in carbohydrates).
Amino and hydroxy acids
In -amino- and -hydroxy acids, the reference groups are NH 2 and OH groups, respectively:

If an amino or hydroxy acid contains several amino or hydroxy groups, then indicate their relative position using the prefixes “erythro”, “threo”, etc. The assignment of an acid to the D- or L-series is determined by the NH 2 or OH group located in the - position to the COOH group located at the top in the Fischer projection:

In this case the letters D and L, indicating the position of the reference group, are provided with the index “S”. This is done to avoid confusion. The index "S" emphasizes that the configuration of the upper chiral center is indicated, which is located in the - position relative to the carboxyl group, as in the amino acid serine ("S" - from the word "serine").

For hydroxy acids with several OH groups, as well as aminohydroxy acids, an alternative configuration designation is used, in which the reference group is the lowest HO group in the Fischer projection. In this case, configuration descriptors D and L are provided with the subscript "g" (from "glyceric aldehyde"). In this case, the amino acids shown in Fig. 123 and 124 are named: D g -threonine (L s - threonine) and L g -threonine ( D s - threonine).
Carbohydrates.
In carbohydrates the reference group is lowest in the Fischer projection, a hydroxyl group bonded to an ammetic carbon atom


It is obvious that in the case of molecules with one asymmetric atom, the D,L nomenclature, like the R,S nomenclature, unambiguously speaks about the absolute configuration of the center of chirality. The same applies to the application of D,L -names of stereoisomers with several asymmetric atoms, since in this case the configuration of the remaining chirality centers is specified by the prefixes erythro-, threo-, ribo-, lyxo-, etc. So, if we say "threose", we will only specify relative configuration of asymmetric atoms in a molecule. Then it will be unclear which enantiomer we are talking about: (26) or (27). If we say “D-threose”, we will clearly indicate that we mean isomer (26), since in it the reference group OH is located on the right in the Fischer projection:

Thus, the name "D-threose" (like "L-threose") refers to the absolute configuration of both asymmetric atoms in the molecule.
Like the R,S nomenclature, the D,L system for naming stereoisomers is not related to the sign of optical rotation.
It should be noted that previously lowercase letters d were used to indicate the direction of rotation of the plane of polarization of light
(right) and l
(left). The use of these letters should not be confused with the use of capital letters D
and L to indicate the molecular configuration. Currently, the direction of rotation of the plane of polarization of light is usually denoted by the symbols (+) and (-).
5. Chiral molecules without asymmetric atoms
In the previous sections, molecules were considered whose chirality is determined by a certain spatial arrangement of four different atoms or groups of atoms relative to a certain center, called the center of chirality.
There may be cases when there are no such centers in the molecule, but nevertheless the molecule is chiral, since it lacks symmetry elements of the Sn group. In such cases, enantiomers differ in the arrangement of atoms
relative to some axis or plane, which is called the chiral axis or chiral plane. The chirality axis is found, for example, in cumulene molecules.
The structure of the molecule of the simplest cumulene - allene - is such that its two CH 2 fragments are located in two mutually perlendicular planes:

The Allene molecule is achiral: it has two planes of symmetry (shown in the figure). The molecules of butadiene-1,2 and 3-methyl-butadiene-1,2 are also achiral

If we look at the pentadiene-2,3 molecule, we will see that it has no planes of symmetry (just as there are no other symmetry elements of the Sn group). This diene exists as a pair of enantiomers:

The chirality of molecules (28) and (29) is due to a certain spatial arrangement of substituents relative to the axis (shown in the figure) passing through the carbon atoms connected by double bonds. This axis is called chirality axis. Molecules like (28) and (29) are said to have axial chirality.
Chirality axes are also present in the molecules of some other compounds, for example, spiro compounds (spirans):

The mentioned anthropoisomers of ortho-disubstituted biphenyls are also molecules with axial chirality. Examples of molecules with plane of chirality molecules of para-cyclophanes can serve:

The enantiomers depicted here cannot transform into each other due to rotation around the -bonds due to the spatial requirements of the fragments included in these molecules.
R,S nomenclature can be used to denote the configuration of molecules with axial and planar chirality. Those interested can find a description of the principles for assigning a configuration to R or S for such molecules in the VINITI publication: IUPAC Nomenclature Rules for Chemistry, vol. 3, semi-volume 2, M., 1983.
6. To the rule of sequence in R, S - nomenclature.
In a number of cases, when determining the order of precedence of deputies, complications arise. Let's consider some of them.
Example 1.
It is obvious that in this case the junior substituents at the asymmetric carbon atom marked with an asterisk are H (d) and CH 3 (c). Let us consider the two remaining complex substituents, arranging their atoms in layers.

In the first layer of both substituents, the atoms are identical. In the second layer, the set of atoms is also the same. (H,C,O). Therefore, we need to turn to the third layer of atoms. In this case, in the left and right substituents one should first of all compare atoms of the III layer associated with senior atoms of the II layer(that is, consider the “senior branches” of both proxies). In this case we are talking about atoms associated with the oxygen atom of the P layer. Since the C atom in the right substituent is bound to the oxygen atom, and the H atom is bound to the oxygen atom in the left substituent, the right substituent receives priority:

The connection should be assigned an R configuration:

If the atoms of the “senior branch” in the third layer turned out to be the same, for example, both C, then it would be necessary to compare the atoms of the same III layer, but in the junior branch. Then the left deputy would have gained an advantage. However, we do not reach this point in our comparisons, since we can make a choice based on the differences in the atoms layer of the older branch.
In a completely similar way, the choice of order of precedence is carried out, for example, between the following substituents:

A situation may arise when, in order to select a senior deputy, it is necessary to “go through” a multiple connection. In this case, resort to the help of the so-called phantom atoms, having zero atomic number (that is, a priori the lowest) and a valency equal to 1.

In this example, nadr make a choice between left- and right-handed carbon-containing substituents. Let us consider them, having previously “opened” the double C=C bond of the first substituent. In this case, duplicated atoms will appear (highlighted in circles). We will add phantom atoms to the duplicate atoms (denote them by the letter f) so as to bring the valence of each to 4:

Now we can compare the left and right substituents:

The difference in the third layer of atoms allows us to give priority to the right-hand substituent:

Therefore, the connection has an R configuration.
Example 3. In some cases, two substituents on an asymmetric atom are structurally identical, but differ only in the absolute configuration of the chiral centers. Then they accept that- R-configuration is older than S-configuration. Accordingly, the central carbon atom in the example below should be assigned the S configuration:

Example 4. The principles outlined above are also applicable to describe the absolute configuration of asymmetric atoms with three substituents (nitrogen, phosphorus, sulfur atoms). In this case, a phantom atom is used as the fourth substituent, which is always the youngest (a lone pair of electrons can be considered as a phantom atom):

Example 5. Sometimes, to select the seniority of substituents, it is necessary to “open” the cycle, just as one “opens” a multiple bond.

In this case, it is easy to determine the most senior (O) and the youngest (H) substituents at the carbon atom marked with an asterisk. In order to make a choice between carbon atoms 1 Cu 2 C, you should “open” the ring along the 2 C-O bond according to the following scheme (duplicate atoms are highlighted in circles):

In this case, in contrast to the “opening” of multiple bonds, duplicated atoms no longer represent “dead-end” branches, but are continued in the repetition of the atom marked with an asterisk. That is, the procedure of “opening” the cycle ends when the same atom (or rather, its duplicate) appears at the ends of both branches. Now we can compare the 1 Cu 2 C atoms by looking at the corresponding layers of atoms:

The difference in the third layer allows for an advantage in seniority - carbon atom 2 C. Consequently, the chiral center in question has an S-configuration:

1.E.Iliel, Fundamentals of Stereochemistry. M.: Mir, 1971, 107 pp.
2.V.M.Potapov, Stereochemistry. M.: Chemistry, 1988, 463 p.
3.V.I.Sokolov, Introduction to theoretical stereochemistry, M., Nauka, 1979, 243 p.
Concept chirality– one of the most important in modern stereochemistry. A model is chiral if it does not have any symmetry elements (plane, center, mirror-rotational axes), except simple axes of rotation. We call a molecule that is described by such a model chiral (meaning “hand-like”, from the Greek . hiro- hand) for the reason that, like hands, molecules are incompatible with their mirror images. In Fig. Figure 1 shows a number of simple chiral molecules. Two facts are absolutely obvious: firstly, the pairs of given molecules represent mirror images of each other, and secondly, these mirror reflections cannot be combined with each other. It will be noted that in each case the molecule contains a carbon atom with four different substituents. Such atoms are called asymmetric. An asymmetric carbon atom is a chiral or stereogenic center. This is the most common type of chirality. If a molecule is chiral, then it can exist in two isomeric forms, related as an object and its mirror image and incompatible in space. Such isomers (para) are called enantiomers.
The term “chiral” does not allow for free interpretation. When a molecule is chiral, then, by analogy with a hand, it must be either left-handed or right-handed. When we call a substance or some sample of it chiral, this simply means that it (it) consists of chiral molecules; Moreover, it is not at all necessary that all molecules are identical in terms of chirality (left or right, R or S, see section 1.3). Two limiting cases can be distinguished. In the first, the sample consists of molecules identical in terms of chirality (homochiral, only R or just S); such a pattern is called enantiomerically pure. In the second (opposite) case, the sample consists of the same number of molecules different in terms of chirality (heterochiral, molar ratio R: S=1:1); such a sample is also chiral, but racemic. There is also an intermediate case - a non-equimolar mixture of enantiomers. This mixture is called scalemic or non-racemic. Thus, the statement that a macroscopic sample (as opposed to an individual molecule) is chiral should be considered not entirely clear and therefore in some cases insufficient. Additional indication may be required whether the sample is racemic or non-racemic. Lack of precision in understanding this leads to a certain kind of misconception, for example, in the titles of articles, when the synthesis of some chiral compound is proclaimed, but it remains unclear whether the author simply wants to draw attention to the very fact of chirality of the structure discussed in the article, or whether the product was actually obtained in the form a single enantiomer (i.e., an ensemble of homochiral molecules; this ensemble, however, should not be called a homochiral sample). Thus, in the case of a chiral non-racemic sample, it is more correct to say "enantiomerically enriched" or " enantiomerically pure".
Methods for depicting optical isomers
The image method is chosen by the author solely for reasons of convenience of conveying information. In Figure 1, images of enantiomers are given using perspective pictures. In this case, it is customary to draw connections lying in the image plane with a solid line; connections going beyond the plane are dotted; and connections directed towards the observer are marked with a thick line. This method of depiction is quite informative for structures with one chiral center. These same molecules can be depicted as a Fischer projection. This method was proposed by E. Fisher for more complex structures (in particular, carbohydrates) having two or more chiral centers.







Mirror plane
Rice. 1
To construct Fischer projection formulas, the tetrahedron is rotated so that two bonds lying in the horizontal plane are directed towards the observer, and two bonds lying in the vertical plane are directed away from the observer. Only the asymmetric atom falls on the image plane. In this case, the asymmetric atom itself is usually omitted, retaining only the intersecting lines and substituent symbols. To remember the spatial arrangement of substituents, a broken vertical line is often preserved in projection formulas (the upper and lower substituents are removed beyond the plane of the drawing), but this is often not done. Below are examples of different ways to depict the same structure with a specific configuration (Fig. 2)
Fischer projection

Rice. 2
Let us give several examples of Fischer projection formulas (Fig. 3)

(+)-(L)-alanine(-)-2-butanol (+)-( D)-glyceraldehyde
Rice. 3
Since the tetrahedron can be viewed from different sides, each stereoisomer can be depicted with twelve (!) different projection formulas. To standardize projection formulas, certain rules for writing them have been introduced. Thus, the main (nomenclatural) function, if it is at the end of the chain, is usually placed at the top, the main chain is depicted vertically.
In order to compare “non-standard” written projection formulas, you need to know the following rules for transforming projection formulas.
1. The formula cannot be removed from the drawing plane and cannot be rotated by 90 o, although it can be rotated in the drawing plane by 180 o without changing their stereochemical meaning (Fig. 4)

Rice. 4
2. Two (or any even number) rearrangements of substituents on one asymmetric atom do not change the stereochemical meaning of the formula (Fig. 5)

Rice. 5
3. One (or any odd number) rearrangement of substituents at the asymmetric center leads to the formula for the optical antipode (Fig. 6)

Rice. 6
4. A rotation in the drawing plane by 90 0 turns the formula into an antipodeal one, unless at the same time the condition for the location of the substituents relative to the drawing plane is changed, i.e. assume that now the lateral substituents are behind the drawing plane, and the upper and lower ones are in front of it. If you use a formula with a dotted line, then the changed orientation of the dotted line will directly remind you of this (Fig. 7)

Rice. 7
5. Instead of permutations, projection formulas can be transformed by rotating any three substituents clockwise or counterclockwise (Fig. 8); the fourth substituent does not change its position (this operation is equivalent to two permutations):

Rice. 8
Fischer projections cannot be applied to molecules whose chirality is related not to the chiral center, but to other elements (axis, plane). In these cases, 3D images are needed.
D , L - Fisher nomenclature
We discussed one problem - how to depict a three-dimensional structure on a plane. The choice of method is dictated solely by the convenience of presenting and perceiving stereo information. The next problem relates to composing a name for each individual stereoisomer. The name should reflect information about the configuration of the stereogenic center. Historically, the first nomenclature for optical isomers was D, L- nomenclature proposed by Fisher. Until the 1960s, it was more common to denote the configuration of chiral centers based on planar projections (Fisher) rather than on the basis of three-dimensional 3D formulas, using descriptors DAndL. Currently D, L– the system is used limitedly - mainly for such natural compounds as amino acids, hydroxy acids and carbohydrates. Examples illustrating its application are shown in Fig. 10.

Rice. 10
For α – amino acids, the configuration is indicated by the symbol L, if in the Fischer projection formula the amino – (or ammonium) group is located on the left; symbol D used for the opposite enantiomer. For sugars, the configuration designation is based on the orientation of the highest-numbered OH group (farthest from the carbonyl end). If the OH group is directed to the right, then this is a configuration D; if HE is on the left – configuration L.
At one time, Fischer's system made it possible to create a logical and consistent stereochemical taxonomy of a large number of natural compounds originating from amino acids and sugars. However, the limitations of the Fischer system, as well as the fact that in 1951 the X-ray diffraction method appeared to determine the true arrangement of groups around the chiral center, led to the creation in 1966 of a new, more rigorous and consistent system for describing stereoisomers, known as R, S - Kahn-Ingold-Prelog nomenclature (KIP). In the instrumentation system, special descriptors are added to the usual chemical name R or S(in italics in the text), strictly and unambiguously defining the absolute configuration.
NomenclatureCana-Ingolda-Preloga
To define a handle R or S for a given chiral center, the so-called chirality rule. Let's consider four substituents connected to the chiral center. They should be arranged in a uniform sequence of stereochemical precedence; for convenience, let's denote these substituents by the symbols A, B, D and E and agree to assume that in the general sequence of precedence (in other words, by priority) A is older than B, B is older than D, D is older than E(A>B>D>E) . The CIP chirality rule requires that the model be considered from the side opposite to that occupied by the substituent E with the lowest priority or the stereochemically junior substituent (Fig. 11). Then the remaining three substituents form something like a tripod, the legs of which are directed towards the viewer.

Rice. eleven
If the seniority of substituents in the row A>B>D falls clockwise (as in Fig. 11), then the center is assigned a configuration descriptor R ( from Latin word rectus - right). In another arrangement, when the stereochemical priority of the substituents decreases counterclockwise, the center is assigned a configuration descriptor S (from Latin sinister - left).
When depicting connections using Fisher projections, the configuration can be easily determined without building spatial models. The formula must be written so that the junior substituent is at the bottom or at the top, since according to the rules for representing Fischer projections, vertical connections are directed away from the observer (Fig. 12). If the remaining substituents are arranged clockwise in decreasing order of precedence, the compound is classified as ( R)-row, and if counterclockwise, then to ( S)-row, for example:

Rice. 12
If the junior group is not on vertical connections, then it should be swapped with the lower group, but remember that this reverses the configuration. You can make any two permutations without changing the configuration.
Thus, the determining factor is stereochemical precedence . Let's discuss now precedence rules, i.e. rules by which groups A, B, D and E are ranked in order of priority.
Preference in terms of seniority is given to atoms with greater atomic number. If the numbers are the same (in the case of isotopes), then the atom with the highest atomic mass becomes older (for example, D>H). The youngest “substituent” is a lone electron pair (for example, in nitrogen). Thus, precedence increases in the series: lone pair
Consider a simple example: in bromochlorofluoromethane CHBrCIF (Fig. 13) there is one stereogenic center, and the two enantiomers can be distinguished as follows. First, substituents are ranked according to their stereochemical seniority: the higher the atomic number, the older the substituent. Therefore, in this example, Br > C1 > F > H, where “>” means “more preferred” (or “older”). The next step is to look at the molecule from the side opposite the youngest substituent, in this case hydrogen. It can be seen that the three remaining substituents are located in the corners of the triangle and are directed towards the observer. If the seniority of this trio of substituents decreases clockwise, then this enantiomer is designated as R. In another arrangement, when the seniority of substituents decreases counterclockwise, the enantiomer is designated as S. Designations R And S write in italics and placed in parentheses before the name of the structure. Thus, the two enantiomers considered have the names ( S)-bromochlorofluoromethane and ( R)-bromochlorofluoromethane.

Rice. 13
2. If two, three or all four identical atoms are directly associated with an asymmetric atom, seniority is established by the atoms of the second belt, which are no longer associated with the chiral center, but with those atoms that had the same seniority.

Rice. 14
For example, in the molecule of 2-bromo-3-methyl-1-butanol (Fig. 14), the highest and youngest substituents are easily determined by the first belt - these are bromine and hydrogen, respectively. But it is not possible to establish seniority based on the first atom of the CH 2 OH and CH(CH 3) 2 groups, since in both cases it is a carbon atom. To determine which group is older, the sequence rule is again applied, but now the atoms of the next belt are considered. Compare two sets of atoms (two triplets), written in order of decreasing precedence. Seniority is now determined by the first point where a difference is found. Group WITH H 2 OH - oxygen, hydrogen, hydrogen WITH(ABOUT NN) or in numbers 6( 8 eleven). Group WITH H(CH 3) 2 – carbon, carbon, hydrogen WITH(WITH CH) or 6( 6 61). The first point of difference is emphasized: oxygen is older than carbon (in atomic number), so the CH 2 OH group is older than CH(CH 3) 2. The configuration of the enantiomer shown in Figure 14 can now be denoted as ( R).
If such a procedure does not lead to the construction of an unambiguous hierarchy, it is continued at increasingly increasing distances from the central atom until, finally, differences are encountered and all four substituents receive their seniority. In this case, any preference acquired by one or another deputy at one of the stages of coordination of seniority is considered final and is not subject to revaluation at subsequent stages.
3. If there are branch points in the molecule, the procedure for establishing the seniority of atoms should be continued along the molecular chain of the highest seniority. Suppose we need to determine the sequence of precedence of the two substituents shown in Fig. 15. It is obvious that the solution will not be achieved either in the first (C), or in the second (C, C, H) or in the third (C, H, F, C, H, Br) layers. In this case, you will have to move to the fourth layer, but this should be done along the path, the advantage of which is established in the third layer (Br > F). Therefore, the decision on the priority of the deputy IN over the deputy A is done on the basis that in the fourth layer Br >CI for that branch, the transition to which is dictated by the seniority in the third layer, and not on the basis that the I atom has the highest atomic number in the fourth layer (which is on the less preferred one and therefore not branch under study).

Rice. 15
4. Multiple connections are represented as the sum of the corresponding simple connections. In accordance with this rule, each atom connected by a multiple bond is assigned an additional “phantom” atom (or atoms) of the same kind located at the other end of the multiple bond. Complementary (additional or phantom) atoms are enclosed in parentheses and are considered to carry no substituents in the next layer. As an example, consider the representations of the following groups (Fig. 16).
Group Presentation

Rice. 16
5. An artificial increase in the number of substituents is also required when the substituent (ligand) is bidentate (or tri- or tetradentate), and also when the substituent contains a cyclic or bicyclic fragment. In such cases, each branch of the cyclic structure is cut after the branch point [where it bifurcates into itself], and the branch point atom is placed (in parentheses) at the end of the chain resulting from the cut. In Fig. 17, using the example of a tetrahydrofuran (THF) derivative, the case of a bidentate (cyclic) substituent is considered. The two arms of the five-membered ring (individually) are cut at bonds with a chiral atom, which is then added to the end of each of the two newly formed chains. It can be seen that as a result of dissection A a hypothetical substituent -CH 2 OCH 2 CH 2 -(C) is obtained, which turns out to be older than the real acyclic substituent -CH 2 OCH 2 CH 3 due to the advantage of the phantom (C) at the end of the first substituent. On the contrary, formed as a result of dissection IN the hypothetical ligand –CH 2 CH 2 OCH 2 – (C) turns out to be lower in seniority than the real substituent –CH 2 CH 2 OCH 2 CH 3, since the latter has three hydrogen atoms attached to the terminal carbon, while the former has none in this layer. Consequently, taking into account the established order of precedence of substituents, the configuration symbol for a given enantiomer turns out to be S.
Determine seniority Deputy A
IN>A
Deputy A

Fig.17

Rice. 18
A similar case of cutting of a cyclic substituent is illustrated by the example of the compound in Fig. 18 where structure IN illustrates the interpretation of the cyclohexyl ring (in the structure A). In this case, the correct precedence sequence is di- n-hesylmethyl > cyclohexyl > di- n-pentylmethyl > N.
Now we are sufficiently prepared to consider such a substituent as phenyl (Fig. 19 structure A). We discussed the scheme for opening each multiple connection above. Since (in any Kekule structure) each of the six carbon atoms is double bonded to another carbon atom, then (in the KIP system) each carbon atom of the ring carries an additional carbon as a “substituent”. The ring supplemented in this way (Fig. 19, structure IN) is then expanded according to the rules for cyclic systems. As a result, the dissection is described by the diagram shown in Fig. 19, the structure WITH.

Rice. 19
6. We will now consider chiral compounds in which the differences between the substituents are not of a material or constitutional nature, but are reduced to differences in configuration. Compounds containing more than one chiral center will be discussed below (see section 1.4). Here we will touch on substituents that differ cis–trans– isomerism (olefin type). According to Prelog and Helmchen, the olefin ligand in which the senior substituent is located on the same side from the double bond of the olefin, which is the chiral center, has an advantage over the ligand in which the senior substituent is in trance–position towards the chiral center. This position has nothing to do with the classical cis-trans-, neither to E–Z–nomenclature for double bond configuration. Examples are shown in Fig. 20.

Rice. 20
Compounds with multiple chiral centers
If a molecule has two chiral centers, then since each center can have (R)- or ( S)-configuration, the existence of four isomers is possible - R.R., SS, R.S. And S.R.:

Rice. 21
Since the molecule has only one mirror image, the enantiomer of the compound is (R.R.) can only be an isomer (SS). Similarly, another pair of enantiomers forms isomers (R.S.) And (S.R.). If the configuration of only one asymmetric center changes, then such isomers are called diastereomers. Diastereomers are stereoisomers that are not enantiomers. So, diastereomeric pairs (R.R.)/(R.S.), (R.R.)/(S.R.), (SS)/(R.S.) And (SS)/(S.R.). Although in general the combination of two chiral centers produces four isomers, the combination of centers of the same chemical structure produces only three isomers: (R.R.) And (SS), being enantiomers, and (R.S.), diastereomeric to both enantiomers (R.R.) And (SS). A typical example is tartaric acid (Fig. 22), which has only three isomers: a pair of enantiomers and meso form.

Rice. 22
meso-Wine acid is (R, S) isomer, which is optically inactive, since the combination of two mirror-symmetric fragments leads to the appearance of a plane of symmetry (a). meso-Wine acid is an example of an achiral compound of meso configuration, which is built from an equal number of chiral elements that are identical in structure but different in absolute configuration.
If the molecule has P chiral centers, the maximum number of stereoisomers can be calculated using formula 2 n; however, sometimes the number of isomers will be less due to the presence of meso forms.
For the names of stereoisomers of molecules containing two asymmetric carbon atoms, two substituents on each of which are the same, and the third are different, prefixes are often used erythro- And trio- from the names of the sugars erythrose and threose. These prefixes characterize the system as a whole, and not each chiral center separately. When depicting such connections using Fischer projections in pairs erythro- isomers, the same groups are located on one side, and if the different groups (C1 and Br in the example below) were the same, the meso form would be obtained. Paired with threo- isomers, the same groups are located on different sides, and if the different groups were the same, the new pair would remain an enantiomeric pair.

Rice. 23
All examples of compounds discussed above have a center of chirality. Such a center is an asymmetric carbon atom. However, other atoms (silicon, phosphorus, sulfur) can also be the center of chirality, as, for example, in methylnaphthylphenylsilane, o-anisylmethylphenylphosphine, methyl p-tolyl sulfoxide (Fig. 24)

Rice. 24
Chirality of molecules lacking chiral centers
A necessary and sufficient condition for the chirality of a molecule is its incompatibility with its mirror image. The presence of a single (configurationally stable) chiral center in a molecule is a sufficient, but not at all necessary, condition for the existence of chirality. Let us consider chiral molecules lacking chiral centers. Some examples are shown in Figures 25 and 26.

Rice. 25

Rice. 26
These are compounds with chirality axes ( axial type of chirality): allenes; alkylidenecycloalkanes; spiranes; so-called atropoisomers (biphenyls and similar compounds, the chirality of which arises due to hindered rotation around a single bond). Another element of chirality is the chirality plane ( planar chirality). Examples of such compounds are ansa compounds (in which the alicyclic ring is too small for the aromatic ring to rotate through); paracyclophanes; metallocenes. Finally, the chirality of a molecule can be related to the helical organization of the molecular structure. The molecule can wrap itself into either a left-handed or right-handed helix. In this case, we talk about helicity (spiral type of chirality).
In order to determine the configuration of a molecule having chirality axis, it is necessary to introduce an additional point into the sequence rule: the groups closest to the observer are considered older than the groups remote from the observer. This addition must be made, since for molecules with axial chirality the presence of identical substituents at opposite ends of the axis is acceptable. Application of this rule to the molecules shown in Fig. 25, shown in Fig. 27.

Rice. 27
In all cases, the molecules are viewed along the chiral axis on the left. It should be understood that if the molecules are considered on the right, then the configuration descriptor will remain the same. Thus, the spatial arrangement of the four support groups corresponds to the vertices of the virtual tetrahedron and can be represented using the corresponding projections (Fig. 27). To determine the appropriate descriptor we use standard rules R, S- nomenclature. In the case of biphenyls, it is important to note that the substituents in the ring are considered from the center (through which the chiral axis passes) to the periphery, in violation of standard sequence rules. So, for biphenyl in Fig. 25 correct sequence of substituents in the right ring C-OSH 3 >C-H; the chlorine atom is too distant to be taken into account. The supporting atoms (those by which the configuration symbol is determined) turn out to be the same if the molecule is viewed from the right. Sometimes descriptors are used to distinguish axial chirality from other types aR And aS (or R a And S a), however the use of the prefix " a» is not mandatory.
Alternatively, molecules with chirality axes can be thought of as helical, and their configuration can be denoted by the symbols R And M. In this case, to determine the configuration, only substituents with the highest priority are considered in both the front and rear (remote from the observer) parts of the structure (substituents 1 and 3 in Fig. 27). If the transition from the highest priority front deputy 1 to the priority rear deputy 3 is clockwise, then this is the configuration R; if counterclockwise, this is the configuration M.
In Fig. 26 shows molecules with planes of chirality. The definition of the chiral plane is not as easy and unambiguous as the definition of the center and axis of chirality. This is a plane that contains as many atoms of the molecule as possible, but not all. In fact, chirality occurs because (and only because) at least one substituent (usually more) does not lie in the plane of chirality. Thus, the chiral plane of the ansa-compound A is the plane of the benzene ring. In paracyclophane IN the most substituted (lower) ring is considered as the chiral plane. To determine a descriptor for planar chiral molecules, a plane is viewed from the side of the atom closest to the plane but not in the plane (if there are two or more candidates, then the one closest to the atom with the highest priority is selected according to the sequence rules ). This atom, sometimes called a test or pilot atom, is indicated by an arrow in Fig. 26. Then, if three consecutive atoms (a, b, c) with the highest priority form a broken line in the chiral plane, bending clockwise, then the configuration of the compound pR (or R p), and if the polyline bends counterclockwise, then the configuration descriptor pS(or S p). Planar chirality, like axial chirality, can alternatively be considered a type of chirality. In order to determine the direction (configuration) of the helix, one must consider the pilot atom together with atoms a, b and c, as defined above. From this it is clear that pR- connections corresponds R-, A pS- connections – M– helicity.
To determine the absolute configuration of the chiral center, the following operations must be performed:
1. Position the chiral center so that the line of sight is directed from the chiral carbon to the junior substituent.
2. In the resulting projection, the three remaining substituents will be located at an angle of 120 o. If the seniority of substituents decreases clockwise- This R-configuration (the following precedence changes are assumed: A > D > B):
If counterclock-wise - S-configuration:

The absolute configuration can be determined using the Fisher formula. To do this, by actions that do not change the Fischer formula, the junior substituent is placed down. After this, the change in seniority of the three remaining deputies is considered. If the seniority of substituents decreases clockwise, this is an R-configuration; if it decreases in the opposite direction, this is an S-configuration. The junior deputy is not taken into account.
Example
Let's consider determining the configuration of chiral centers using the example of 3-bromo-2-methyl-2-chlorobutanol-1, which has the following structure:
Let us determine the absolute configuration of C 2. To do this, let’s imagine C 3 and C 4, as well as everything associated with them in the form of a radical A:

Now the original formula will look like this:
We determine the seniority of substituents (from senior to junior): Cl > A > CH 2 OH > CH 3 . We make an even number of rearrangements (this does not change the stereochemical meaning of the formula!) so that the lowest substituent is at the bottom:

Now let’s consider the top three substituents in the Fischer formula at the chiral center C2:
It can be seen that the traversal of these substituents in decreasing order of seniority occurs counterclockwise, hence the configuration of this chiral center is S.
We will perform similar actions for another chiral center associated with C 3 . Let's imagine again, this time C 2 and everything connected with it, in the form of a radical IN:
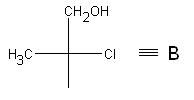
Now the original formula will look like this:
Again, we determine the seniority of the substituents (from senior to junior): Br > B > CH 3 > H. We make an even number of permutations so that the youngest substituent is again at the bottom:

Let us determine in which direction the decrease in seniority occurs (we do not take into account the bottom, youngest deputy!):
The decrease in the seniority of substituents occurs counterclockwise, therefore the configuration of this chiral center is S.
The name of the starting substance, taking into account the absolute configuration of the chiral centers - 3-/S/-bromo-2-/S/-methyl-2-chlorobutanol-1